De kern
-
Wanneer 1 of beide ouders van een patiënt afkomstig zijn uit een malariagebied kan de patiënt drager zijn van hemoglobinopathie.
-
Dragerschap van hemoglobinopathie is geen ziekte, maar een eigenschap die ooit tot doel had malaria te overleven in subtropische gebieden in de wereld.
-
Indien ijzergebrek het microcytair hypochrome bloedbeeld niet verklaart, kan er sprake zijn van thalassemie.
-
Voor onderzoek naar dragerschap van hemoglobinopathie kunt u terecht bij een lokaal klinisch chemisch- of huisartsenlaboratorium of hemoglobinopathie-expertisecentrum.
-
Bespreek partneranalyse indien er een kinderwens is. Wanneer de partner ook drager is, verwijst u naar een klinisch genetisch centrum.
Jaarlijks worden er 35 tot 40 baby’s met een ernstige vorm van hemoglobinopathie geboren, zo blijkt uit cijfers van de hielprikscreening (RIVM-draaiboek hielprikscreening). 1 Dat lijkt misschien niet veel, maar dit evenaart het aantal baby’s met cystische fibrose. 2 Toch is hemoglobinopathie minder bekend dan cystische fibrose. 3
In deze bijdrage geven we een overzicht van de verschillende vormen van hemoglobinopathie en dragerschap, het bijbehorende klinische beeld en het voorkomen in Nederland. Daarnaast doen we handreikingen om diagnostiek aan te vragen, waar ook de prenatale diagnostiek voor deze aandoeningen wordt verricht. 4 Ook laten we zien wat u een risicopaar adviseert en wanneer u naar een klinisch genetisch centrum verwijst. Zo willen we tijdige detectie in de eerstelijnszorg bevorderen.
Hemoglobinopathie
Hemoglobine is een tetrameer eiwit dat bestaat uit 2 alfa- en 2 non-alfa-globineketens (beta of delta). Bij volwassenen bevatten de erythrocyten 97,5% hemoglobine A (alfa2bèta2) en 2,5% hemoglobine A2 (alfa2delta2). Hemoglobinopathieën zijn erfelijke structurele of synthesedefecten van het hemoglobine-eiwit, veroorzaakt door DNA-veranderingen op de alfa- en bètaglobinegenen (zie voor meer informatie: https://hbpinfo.com/). 5 Dat er specifieke genetische defecten in de verschillende populaties voorkomen, is vaak het gevolg van positieve selectie doordat dragers beter in staat zijn om een infectie met malaria tropica te overleven. 6 Hemoglobinopathieën zijn autosomaal recessief overervende hemolytische anemieën. Mannen en vrouwen hebben dus evenveel kans. De ziekte openbaart zich als de eigenschap van beide ouders is overgeërfd.
De meestvoorkomende hemoglobinopathieën zijn thalassemie en sikkelcelziekte. Daarnaast zijn er meer dan 1200 (zoals HbC, HbD, soms zeer zeldzame) hemoglobinevarianten bekend, veroorzaakt door andere DNA-mutaties in de alfa- of bètaglobinegenen. 7 – 9
Dragerschapsfrequentie
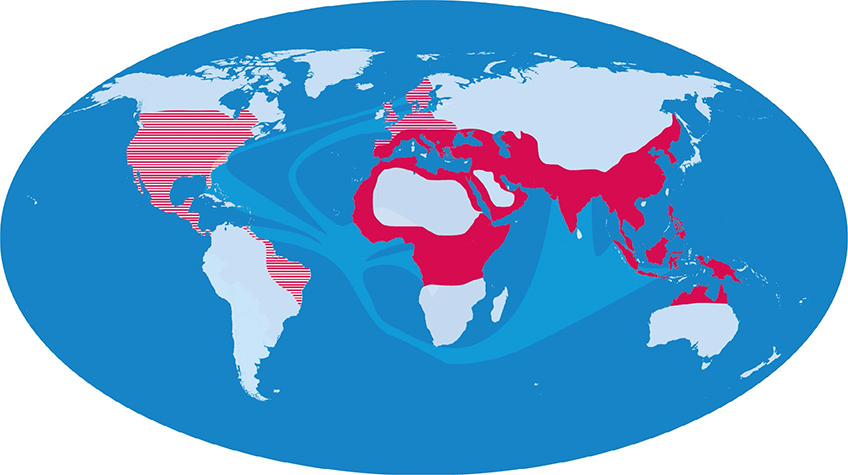
Hemoglobinopathieën behoren tot de meestvoorkomende monogene autosomaal recessieve aandoeningen bij de mens. 11 , 12 Door migratie uit hoogrisicogebieden voor malaria (zoals Centraal- en Noord Afrika, het Nabije en Verre Oosten zoals Indonesië, het Middellandse Zeegebied, Zuid-Amerika en het Caribisch gebied) is het aantal dragers in Noord-Europese landen de afgelopen halve eeuw toegenomen [figuur]. In Nederland wonen ongeveer 2 miljoen mensen afkomstig uit malariagebieden, van wie er naar schatting meer dan 200.000 drager zijn. Door partnerkeuze binnen de familie of etnische groep is het risico op een kind dat is aangedaan met sikkelcelziekte, HbH-ziekte of bètathalassemie major verhoogd. 13 , 14
Figuur | Het geografisch voorkomen van malaria tropica in de wereld en de overlap met hemoglobinopathieën is in rood aangegeven. Door migratie door de eeuwen heen is hemoglobinopathie verspreid naar voorheen niet-endemische gebieden, die gestreept zijn weergegeven.
De populatie patiënten met sikkelcelziekte en thalassemie major is de laatste 16 jaar met ruim 600 gevallen toegenomen. Daarnaast zijn er minstens 12.800 dragers van HbS (sikkelcel-trait) bijgekomen. 15 De werkelijke aantallen patiënten en dragers in de Nederlandse bevolking liggen aanzienlijk hoger. 16 Dat er een brede kloof bestaat tussen het aantal geschatte en het aantal geregistreerde dragers bleek uit een recent onderzoek. 3 , 17 Op basis van dragerschapsfrequentie in de bevolking uit hoogrisicogebieden voor hemoglobinopathie ligt het aantal dragers in de regio Den Haag-Leiden-Delft volgens schattingen op 13.704, terwijl de ELAN-database slechts 1542 dragers met ICPC-code sikkelcelziekte of thalassemie bevat.
Beschouwing
Sikkelcelziekte of bètathalassemie major kunnen worden voorkomen door dragers tijdig te diagnosticeren en eventuele familieleden door te verwijzen voor verder onderzoek en behandeling. Dragers van alfa- en/of bètathalassemie zijn in het algemeen niet ziek, maar kunnen wel een iets afwijkend bloedbeeld hebben, met name een milde microcytair hypochrome anemie. Dragers van HbS hebben een normaal bloedbeeld, tenzij er ook sprake is van alfathalassemie. 5 , 10
De afgelopen jaren is de diagnostiek in Nederland voor hemoglobinopathieën sterk verbeterd en bieden de meeste perifere klinisch chemische laboratoria en huisartsenlabs deze breed aan. 18 Toch wordt dragerschapsdiagnostiek maar sporadisch aangevraagd en krijgt de patiënt bij een incidentele vaststelling van dragerschap onvoldoende of geen genetische informatie, waardoor partneranalyse of familieonderzoek achterwege blijft. 3 , 14
Het verstrekken van informatie over de genetische risico’s van dragerschap en de mogelijkheid tot partneranalyse is bij een kinderwens echter essentieel voor een autonome beslissing over het krijgen van kinderen. Geïnformeerde risicoparen kunnen een verwijzing krijgen naar een klinisch genetisch centrum voor counseling. Daar krijgen ze informatie over prenatale diagnostiek en zwangerschapsafbreking, embryoselectie, het afzien van kinderen of adoptie, of over de manier waarop ze zich kunnen voorbereiden op een ernstige ziekte van het kind. Risicoparen krijgen begeleiding bij het nemen van een weloverwogen beslissing over het nageslacht. Deze werkwijze is ook beschreven in de onlangs verschenen richtlijn van de Vereniging Klinische Genetica Nederland. 19
In Nederland is geen dragerschapsonderzoek naar hemoglobinopathie in het kader van preconceptie- of antenatale screening. Antenatale screening zoals wordt toegepast in het Verenigd Koninkrijk zou als voordeel hebben dat de schaal in Nederland zich beperkt tot de 180.000 zwangerschappen per jaar, waarvan ongeveer 40.000 zwangerschappen binnen de hoogrisicogroep voor hemoglobinopathie vallen. 13 , 21
Thalassemieën
De meestvoorkomende thalassemieën zijn alfa- en bètathalassemie. Het volwassen hemoglobine A (HbA) bestaat uit twee alfa- en twee bètaglobineketens. Iedereen heeft vier alfagenen (twee op elk chromosoom 16) en twee bètagenen (1 op elk chromosoom 11) die verantwoordelijk zijn voor de synthese van de gelijknamige globineketens. Het tekort aan globinesynthese door een genetisch defect van de alfa- of bètagenen wordt respectievelijk alfathalassemie en bètathalassemie genoemd.
Bij alfathalassemie zijn er hetero-, homo- en samengesteld heterozygoten te onderscheiden die naarmate er minder functionele alfagenen zijn een toenemende klinische ernst vertonen. Wanneer de functie van een van beide alfagenen op 1 allel ontbreekt door een deletie wordt dit weergegeven als -α. Als de deletie beide alfagenen wegneemt als - -.. De minst klinisch relevante vormen zijn (-α/αα), (-α/- α) en (- -/αα), die worden aangeduid als dragers. De klinisch relevante vorm (-α/- -) wordt gekenmerkt door matig tot ernstige hemolytische anemie (HbH-ziekte) en de meest ernstige vorm van alfathalassemie is het letale Hb Bart’s hydrops foetalis syndroom. Dit wordt veroorzaakt door de totale afwezigheid van alfagenen (- - /- - ). In verouderde literatuur wordt nog wel gesproken van type 1 alfathalassemie bij het ontbreken van 2 van de 4 alfagenen en van type 2 wanneer 1 alfagen ontbreekt.
Ook bij bètathalassemie bestaan verschillen in ernst. Wanneer de expressie van één van beide bètagenen is verminderd (bèta+) of geheel ontbreekt (bèta0) spreekt men van een drager (bètathalassemie-trait). Bij combinaties van twee bètagen-defecten kan matige tot ernstige bloedarmoede ontstaan (bètathalassemie intermedia) tot zeer ernstige bloedtransfusieafhankelijke vorm van bloedarmoede (bètathalassemie major genaamd).
Sikkelcelziekte
Sikkelcelziekte wordt veroorzaakt door de meestvoorkomende hemoglobinevariant HbS, waarbij op de bètaglobineaminozuurketen een glutaminezuur is vervangen door een valine. Sikkelcelziekte is een multi-orgaanziekte die wordt gekenmerkt door pijnlijke crises, veroorzaakt door occlusie van de haarvaten, zuurstoftekort en schade aan weefsels en organen. De aandoening kan meer of minder ernstig zijn, al naar gelang de gecontinueerde expressie van de foetale gammaglobinegenen in de postnatale periode.
Dragerschapdiagnostiek door de huisarts
Vanwege gebrek aan een screeningsprogramma is dragerschapsonderzoek aangewezen in de huisartsenpraktijk. Indicaties voor dragerschapsonderzoek hiervoor zijn:
-
patiënten in de reproductieve leeftijd met een kinderwens of zwangeren
-
wanneer 1 of beide ouders afkomstig is uit een gebied met een hoge dragerschapfrequentie van hemoglobinopathie
-
bij een hemoglobinopathie bij eerste- of tweedegraads familieleden
-
ouders en familie van een kind bij wie met de hielprik (dragerschap van) een hemoglobinopathie is vastgesteld
-
patiënten met een aanhoudende anemie ondanks ijzersuppletie
De huisarts kan hemoglobinopathie-dragerschaponderzoek bij de meeste lokale (huisartsen)laboratoria aanvragen. Bespreek met de patiënt in het geval van dragerschap de mogelijkheid tot partneranalyse, zeker indien er een kinderwens is. Wanneer de partner ook drager is, verwijs de patiënt naar klinisch genetisch centrum voor verdere begeleiding.
Conclusie
De rol van de huisarts bij het herkennen van dragers uit hoogrisicogroepen voor hemoglobinopathie is cruciaal voor tijdige detectie. Dragerschapsdiagnostiek is van belang voor het bepalen van het genetisch risico op ernstige sikkelcelziekte en thalassemie major, zodat risicoparen tijdig (bij voorkeur vóór de zwangerschap) een verwijzing krijgen naar een klinisch geneticus voor counseling.
Reacties
Er zijn nog geen reacties.